|
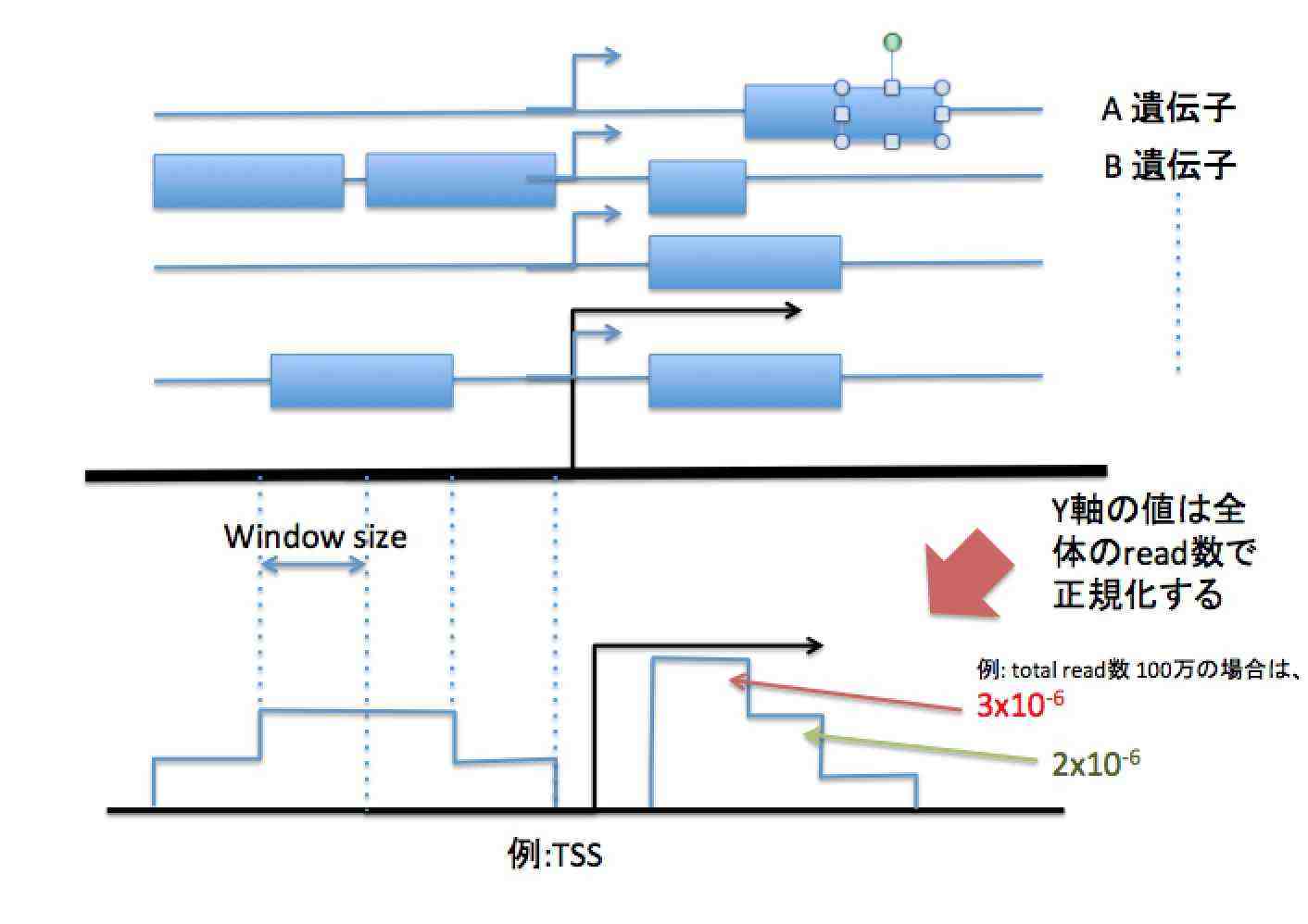
論文のChIP-seq結果をまとめたfigureの解釈に関する質問です。 自分ではChIP-seqを行ったことがなく、データ処理についての経験がありません。 具体的にはhistoneのChIP-seqを行い、転写開始点(TSS)前後におけるhistone variantの分布を示したという図について縦軸(normalized reads; RRM)の値をどう解釈してよいのかが分かりません。 本分からざっくりと意味はわかり、グラフのピークが示す山型を見ればよいと思うのですが、実際にどういう処理をして縦軸がだされたのか、その値は何を意味するのかが理解できません。 こういうグラフを描くことについての計算方法や解釈を示した論文がありましたらご教示いただけると大変ありがたいです。 よろしくお願いいたします。 以下に具体的な例を挙げました。 PubMed Centralでfree accessできる論文ですと、PMC3125718のFigure 1のような図です。 |
|
こんばんは。 suimye様、 ご回答ありがとうございました。 おかげさまでpeak regionのread数分布の作り方は理解することができました。 以下の用に解釈したのですが、正しいでしょうか。 例に挙げた論文のFigure1-aでHighの赤いラインはTSSの少し上流でy軸の値がほぼ1E-8です。 Supplement figureにH2A.Zの総tag数は2.1E+7とあります。 ということは、Highに分類される遺伝子のTSS直上にmappingされるリード数をXとすると、 X/2.1E+7=1E-8で、X=0.21 High遺伝子1000個のうち、0.21個がTSS直上にピークを示す遺伝子である。 ということでしょうか? 確かに黒のsilentや青のmediumのラインと比べれば確かに差はありますが、すごくまれな現象を観察している印象を受けてしまいます。 TSさん、こんにちは。 >High遺伝子1000個のうち、0.21個がTSS直上にピークを示す遺伝子である。
というのは間違いで、 1000個の遺伝子のあるゲノム領域をみると、合計で0.21個のread数
がみられる、となります。 |